

PhD Position available!
The CRECK Modeling Lab is looking for candidates for a Ph.D. position for a research project in collaboration with Brembo S.p.A., a leading company working in the field of disk brake technology. The project is about the numerical modeling of the Chemical Vapor Infiltration (CVI) process for the production of carbon disk brakes for aircraft and racing
New paper on isolated fuel droplets
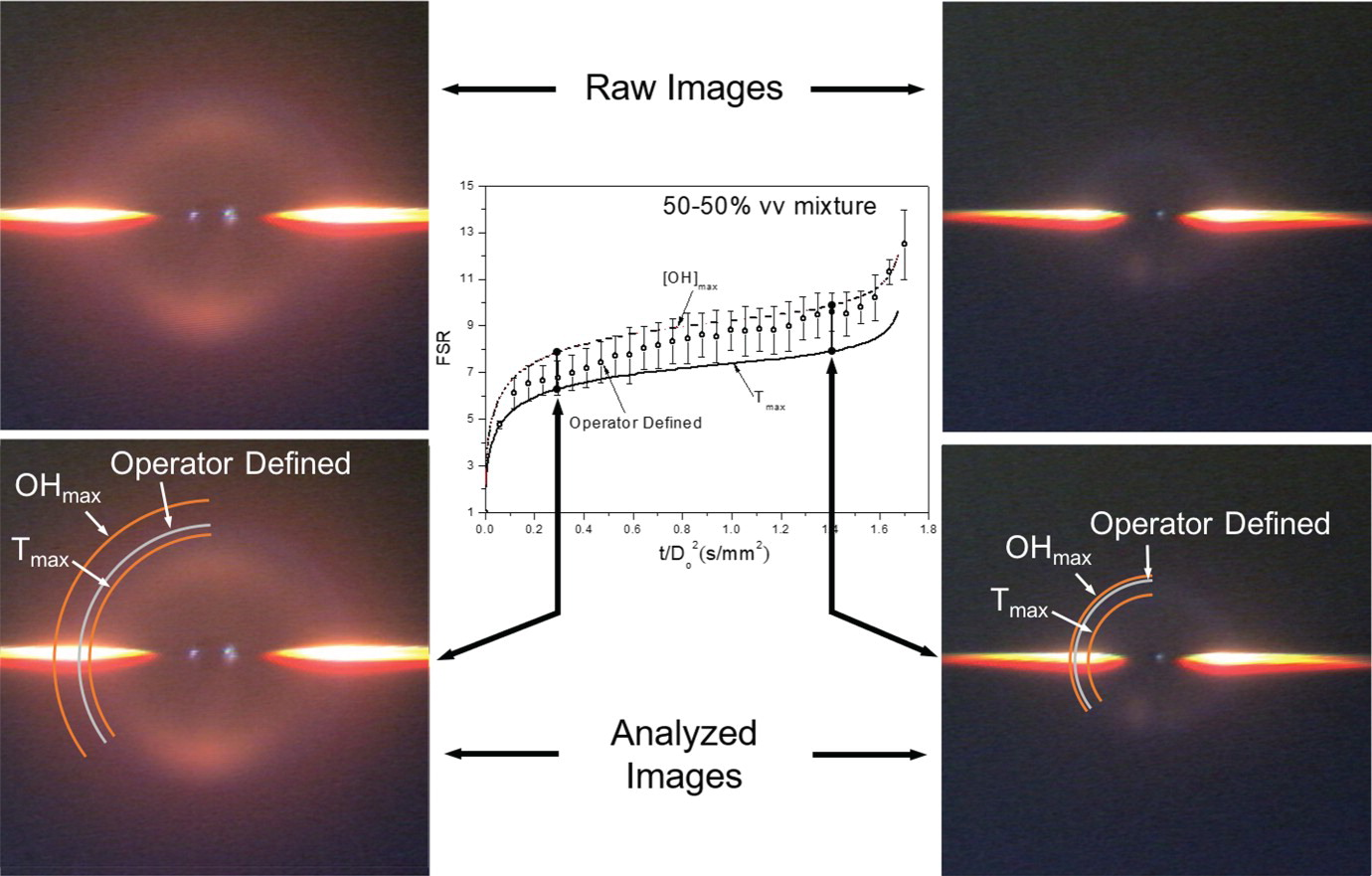
OpenSMOKE++ was used for simulating the evaporation and combustion of isolated fuel droplets. The work is a collaboration between the CRECK Modeling Lab, Cornell University, University of San Diego California and NASA Glenn Research Center. The main results have been published on Combustion Theory and Modelling in a paper with title: “The
New paper on adaptive reduced chemistry of multidimensional flames
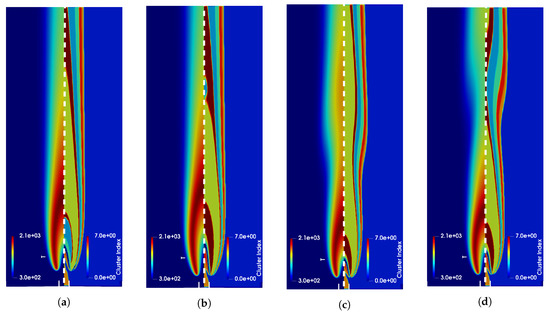
The OpenSMOKE++ framework was used in the context of adaptive reduced chemistry for multidimensional laminar flames in a recent publication on Energies with title: “Impact of the partitioning method on multidimensional adaptive-chemistry simulations”. The work is a collaboration between the CRECK Modeling Lab and the BURN Group at Universite’ Libre de Bruxelles. G. D’Alessio,
New paper on NOx formation in laminar flames
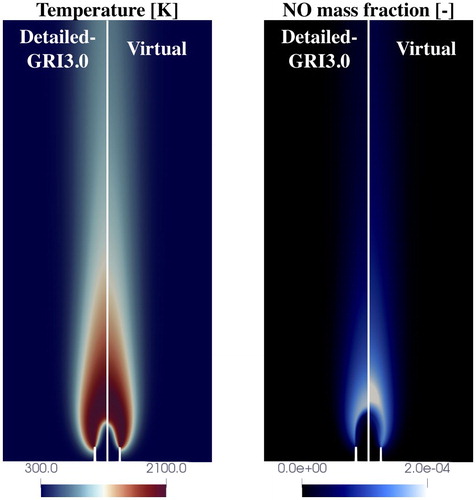
Congratulations to our PhD Candidate Giampaolo Maio! His paper with title “A virtual chemical mechanism for prediction of NO emissions from flames” has been recently published on Combustion Theory and Modelling. G. Maio, M. Cailler, A. Cuoci, B. Fiorina, A virtual chemical mechanism for prediction of NO emissions from flames, (2020), Combustion Theory and
New paper on laminar sooting flames
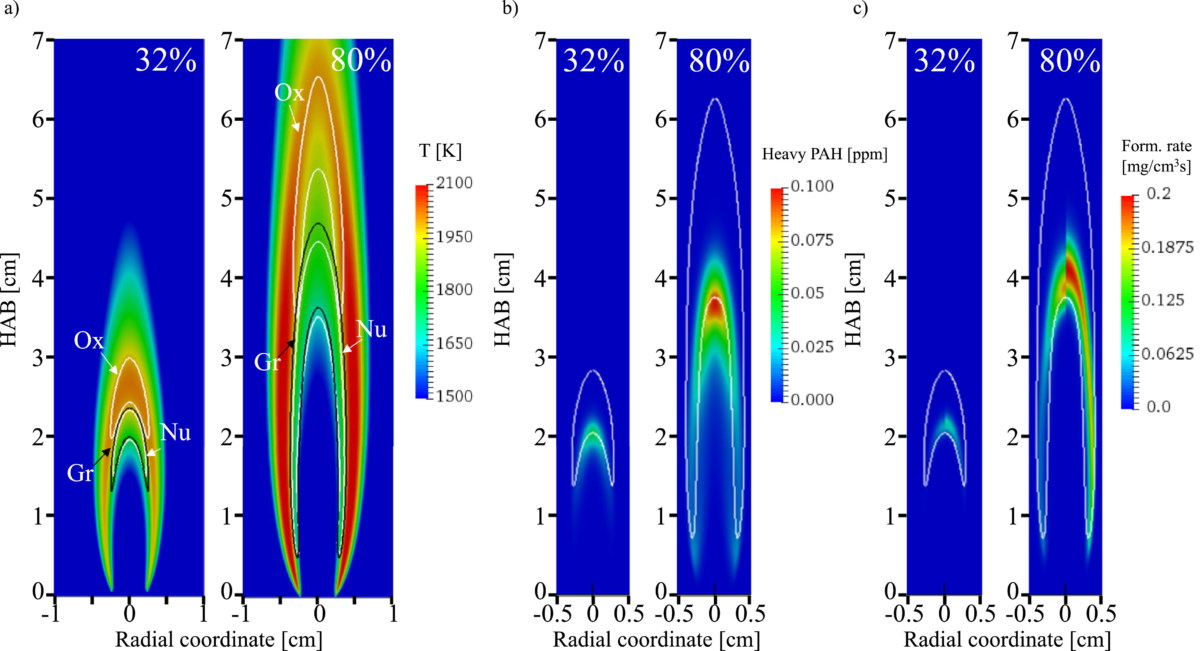
Congratulations to our PhD Candidate Agnes Livia Bodor! Her paper with title “A post processing technique to predict primary particle size of sooting flames based on a chemical discrete sectional model: Application to diluted coflow flames” has been recently published on Combustion and Flame. A.L. Bodor, B. Franzelli, T. Faravelli, A.Cuoci, A post

New paper on droplet evaporation in buoyancy-driven convection
Congratulations to our PhD Candidate Abd Essamade Saufi (aka Sangi)! His paper with title “An experimental and CFD modeling study of suspended droplets evaporation in buoyancy driven convection” has been recently published on Chemical Engineering Journal. A.E.Saufi, R.Calabria, F.Chiariello, A.Frassoldati, A.Cuoci, T.Faravelli, P.Massoli, An experimental and CFD modeling study of suspended droplets evaporation
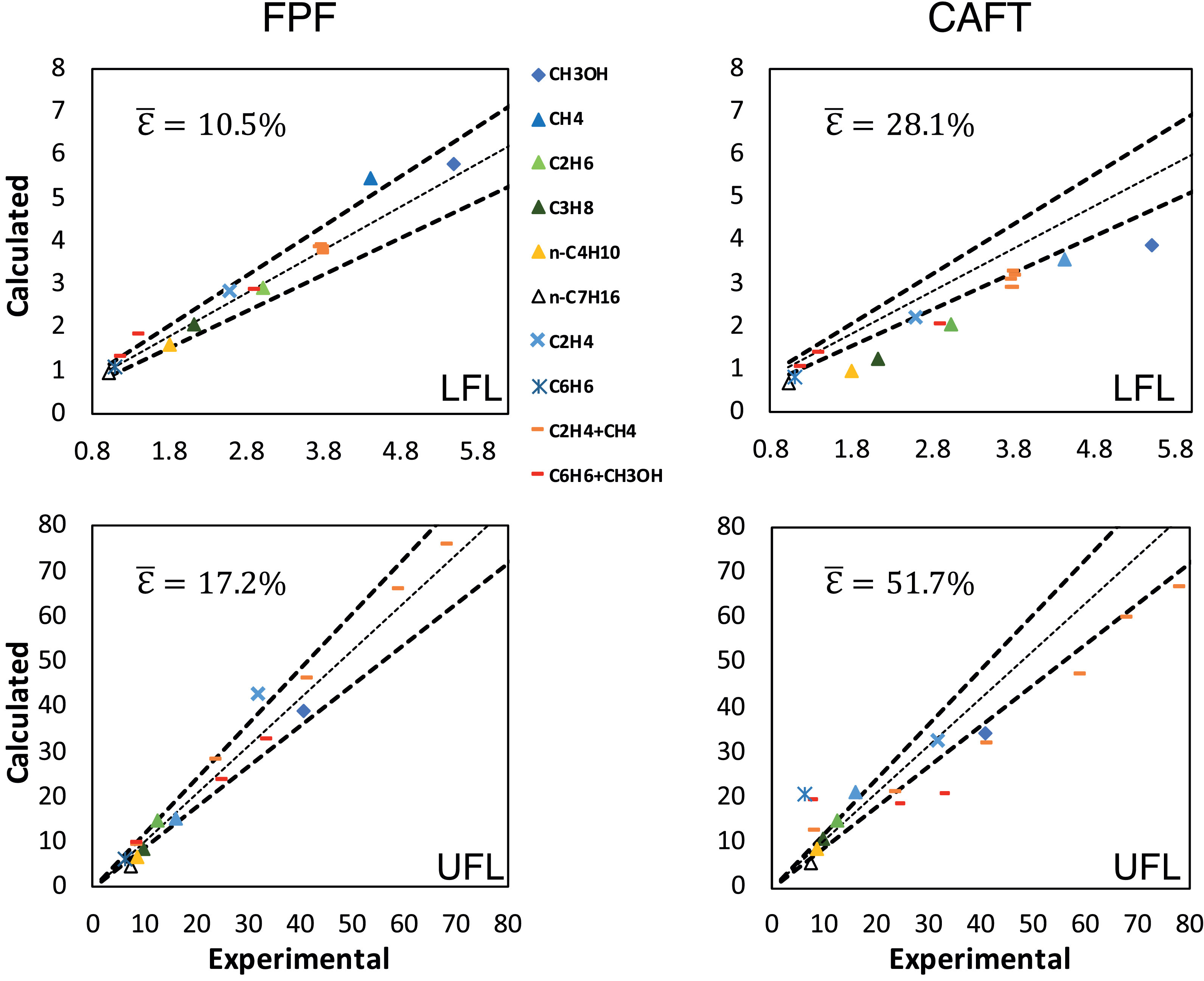
New paper on flammable ranges of hydrocarbons
Congratulations to our PhD Candidate Andrea Bertolino! His paper with title “Prediction of flammable range for pure fuels and mixtures using detailed kinetics” has been recently published on Combustion and Flame. Bertolino A., Stagni A., Cuoci, A., Faravelli, T., Parente A., Frassoldati A., Prediction of flammable range for pure fuels and mixtures using
CRECK at 17th ICNC in Aachen
The CRECK Modeling Lab is proud to have 8 presentations at the Seventeenth International Conference on Numerical Combustion in Aachen (Germany), 6-8 May 2019
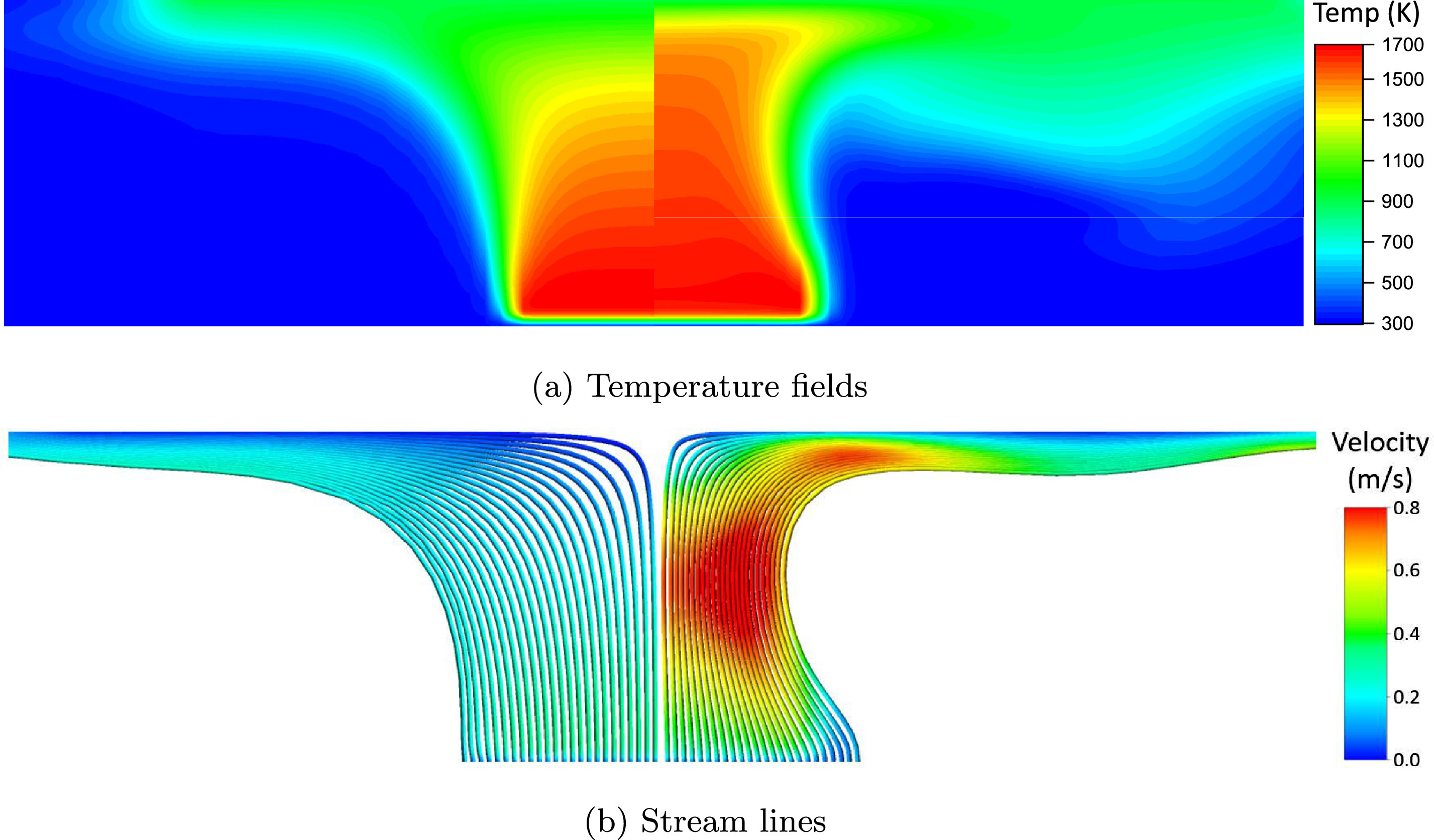
New paper on sooting laminar flames
Congratulations to our PhD Candidate Warumporn Pejpichestakul! Her paper with title “Buoyancy effect in sooting laminar premixed ethylene flame” has been recently published on Combustion and Flame. Pejpichestakul W., Cuoci, A., Frassoldati, A., Pelucchi, M., Parente, A., Faravelli, T., Buoyancy effect in sooting laminar premixed ethylene flame, (2019) Combustion and Flame, 205, pp. 135-146, DOI: 10.1016/j.combustflame.2019.04.001 Abstract

Cover paper on Reaction Chemistry & Engineering
Congratulations to Matteo Pelucchi! His recent paper on “Detailed kinetics of substituted phenolic species in pyrolysis bio-oils” has been selected as the front cover paper of last issue of Reaction Chemistry & Engineering journal. Pelucchi, M., Cavallotti, C., Cuoci, A., Faravelli, T., Frassoldati, A., Ranzi, E. , Detailed kinetics of substituted phenolic species in
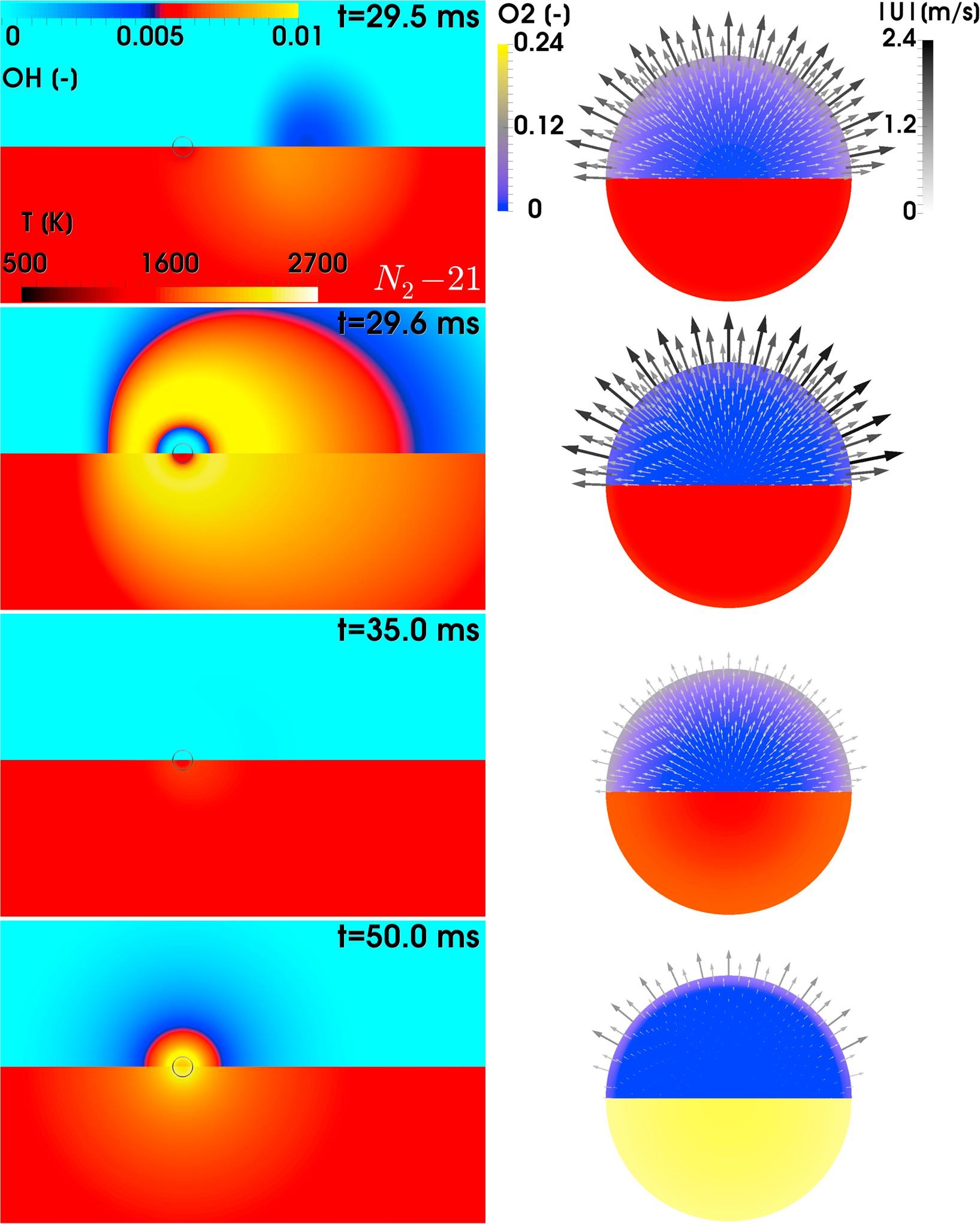
Collaboration with University of Stuttgart
A paper on coal with title “Fully-resolved simulations of coal particle combustion using a detailed multi-step approach for heterogeneous kinetics” has been recently published on Fuel as the result of a successfull collaboration between CRECK Modeling Lab, University of Stuttgart, University of Duisburg Essen, Technical University Freiberg, and Darmstadt University of Technology.
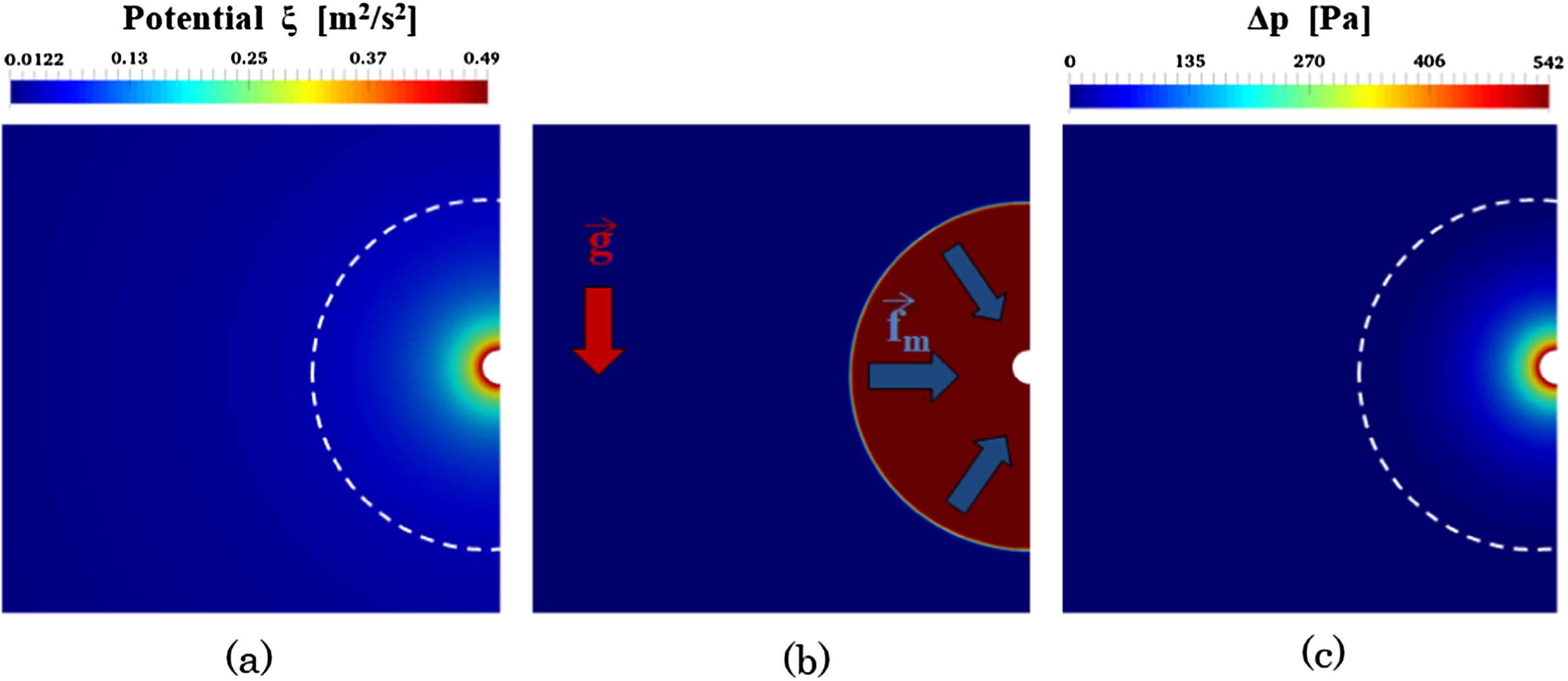
New paper on evaporation of fuel droplets
Congratulations to our PhD Candidate Abd Essamade Saufi! His paper with title “DropletSMOKE++: A comprehensive multiphase CFD framework for the evaporation of multidimensional fuel droplets” has been recently published on International Journal of Heat and Mass Transfer. Saufi, A.E., Frassoldati, A., Faravelli, T., Cuoci, A., DropletSMOKE++: A comprehensive multiphase CFD framework for the evaporation
